How to load your own data & Computing in different genome coordinates & Binding pattern of specific gene!#
the rule is the same. You are just feeding the list of genes in that GO pathway into metadensity and compute them
[1]:
# set up files associated with each genome coordinates
# you need to edit your own .ini files to point the gencode annotation files
import metadensity as md
md.settings.from_config_file('/home/hsher/Metadensity/config/hg19.ini')
# then import the modules
from metadensity.metadensity import *
from metadensity.plotd import *
import pandas as pd
import matplotlib.pyplot as plt
# I have a precompiles list of ENCODE datas as a csv that loads in this dataloader
import sys
sys.path.append('/home/hsher/Metadensity/scripts')
from dataloader import *
%matplotlib inline
plt.style.use('seaborn-white')
Using /projects/ps-yeolab3/bay001/annotations/hg19/hg19.fa
using /projects/ps-yeolab3/bay001/annotations/hg19/hg19.fa
Using: /home/hsher/gencode_coords/gencode.v19.transcript.gff3
Creating eCLIP object for your data#
the eCLIP object needs a pd.Series that stores all the types of files
[2]:
base_dir = '/home/hsher/jackie_ythdf2/'
from metadensity.readdensity import ReadDensity
def gather_data(cell_line):
# store where all the files are
data = pd.Series()
# iterate over specific naming principle.
for rep, rname in zip([1,2], ['R1', 'R2']):
for seq in ['CLIP', 'INPUT']:
# ===== BIGWIGS ======
pos = '{}YTHDF2_trackhubs/hg19/YTHDF2.{}_{}_{}.umi.r1TrTr.sorted.STARUnmapped.out.sorted.STARAligned.outSo.rmDupSo.norm.{}.bw'.format(
base_dir, seq, cell_line,rname, 'pos')
neg = '{}YTHDF2_trackhubs/hg19/YTHDF2.{}_{}_{}.umi.r1TrTr.sorted.STARUnmapped.out.sorted.STARAligned.outSo.rmDupSo.norm.{}.bw'.format(
base_dir, seq, cell_line,rname, 'neg')
if seq == 'CLIP':
data[f'minus_{rep-1}'] = neg
data[f'plus_{rep-1}'] = pos
if seq == 'INPUT':
data[f'minus_control_{rep-1}'] = neg
data[f'plus_control_{rep-1}'] = pos
# ===== BAMS ======
bam = '{}YTHDF2.{}_{}_{}.umi.r1TrTr.sorted.STARUnmapped.out.sorted.STARAligned.outSo.rmDupSo.bam'.format(
base_dir, seq, cell_line, rname)
if seq == 'CLIP':
data[f'bam_{rep-1}'] = bam
if seq == 'INPUT':
data[f'bam_control_{rep-1}'] = bam
# ===== INFORMATION =====
# names
data['uid']=cell_line # THIS THING NEEDS TO BE UNIQUE TO EACH ECLIP LIBRARY. it is called "Unique ID" (uid)
# if uid is NOT UNIQUE it will RUIN EVERYTHING!
data['RBP']=cell_line # name for display when plotting. You can write whatever there. won't interfere computation
# ==== PEAKS ====
if seq == 'CLIP':
fname = '{}YTHDF2.{}_{}_{}.umi.r1TrTr.sorted.STARUnmapped.out.sorted.STARAligned.outSo.rmDupSo.peakClusters.normed.bed'.format(
base_dir, seq, cell_line, rname)
data[f'bed_{rep-1}'] = fname
# Intersect peaks
fname = '{}idr_peaks/{}_1-{}_2.idr.final.bed'.format(base_dir, cell_line, cell_line)
data['idr'] = fname
return data
[3]:
#For reference LM2 and MDA samples are MYC-dependent, MCF7 and SKBR3 are MYC independent
# these data are for YTHDF2 CLIP in different cell lines
lm2 = gather_data('LM2')
mda = gather_data('MDA')
mcf7 = gather_data('MCF7')
SKBR3 = gather_data('SKBR3')
/home/hsher/miniconda3/envs/metadensity/lib/python3.7/site-packages/ipykernel_launcher.py:5: DeprecationWarning: The default dtype for empty Series will be 'object' instead of 'float64' in a future version. Specify a dtype explicitly to silence this warning.
"""
[4]:
lm2 # how the Series should look like
[4]:
minus_0 /home/hsher/jackie_ythdf2/YTHDF2_trackhubs/hg1...
plus_0 /home/hsher/jackie_ythdf2/YTHDF2_trackhubs/hg1...
bam_0 /home/hsher/jackie_ythdf2/YTHDF2.CLIP_LM2_R1.u...
uid LM2
RBP LM2
bed_0 /home/hsher/jackie_ythdf2/YTHDF2.CLIP_LM2_R1.u...
minus_control_0 /home/hsher/jackie_ythdf2/YTHDF2_trackhubs/hg1...
plus_control_0 /home/hsher/jackie_ythdf2/YTHDF2_trackhubs/hg1...
bam_control_0 /home/hsher/jackie_ythdf2/YTHDF2.INPUT_LM2_R1....
minus_1 /home/hsher/jackie_ythdf2/YTHDF2_trackhubs/hg1...
plus_1 /home/hsher/jackie_ythdf2/YTHDF2_trackhubs/hg1...
bam_1 /home/hsher/jackie_ythdf2/YTHDF2.CLIP_LM2_R2.u...
bed_1 /home/hsher/jackie_ythdf2/YTHDF2.CLIP_LM2_R2.u...
minus_control_1 /home/hsher/jackie_ythdf2/YTHDF2_trackhubs/hg1...
plus_control_1 /home/hsher/jackie_ythdf2/YTHDF2_trackhubs/hg1...
bam_control_1 /home/hsher/jackie_ythdf2/YTHDF2.INPUT_LM2_R2....
idr /home/hsher/jackie_ythdf2/idr_peaks/LM2_1-LM2_...
dtype: object
[5]:
# it's worthwhile to see if the data path is correct
import os
lm2.apply(os.path.isfile) # ok it's good to learn every path exists! yeee ha
[5]:
minus_0 True
plus_0 True
bam_0 True
uid False
RBP False
bed_0 True
minus_control_0 True
plus_control_0 True
bam_control_0 True
minus_1 True
plus_1 True
bam_1 True
bed_1 True
minus_control_1 True
plus_control_1 True
bam_control_1 True
idr True
dtype: bool
[6]:
clips = []
# then you build eCLIP from each pd.Series!
for data in [lm2, mda, mcf7, SKBR3]:
clips.append(eCLIP.from_series(data, single_end = True))
Build Metagene for MYC only#
[7]:
myc = transcript.filter(lambda x: x.attrs['gene_name']=='MYC').saveas()
print(myc)
chr8 HAVANA transcript 128748330 128753674 . + . ID=ENST00000377970.2;Parent=ENSG00000136997.10;gene_id=ENSG00000136997.10;transcript_id=ENST00000377970.2;gene_type=protein_coding;gene_status=KNOWN;gene_name=MYC;transcript_type=protein_coding;transcript_status=KNOWN;transcript_name=MYC-001;level=2;protein_id=ENSP00000367207.2;tag=basic,CCDS;ccdsid=CCDS6359.2;havana_gene=OTTHUMG00000128475.3;havana_transcript=OTTHUMT00000250277.3;
[8]:
all_meta_raw = []
for e in clips:
m = Metadensity(e, name = e.name, transcripts = myc,
background_method = None,
normalize = None) # raw read coverage in RPM
m.get_density_array()
all_meta_raw.append(m)
Using: /home/hsher/Metadensity/metadensity/data/hg19/gencode
Done building metagene
Using: /home/hsher/Metadensity/metadensity/data/hg19/gencode
Done building metagene
Using: /home/hsher/Metadensity/metadensity/data/hg19/gencode
Done building metagene
Using: /home/hsher/Metadensity/metadensity/data/hg19/gencode
Done building metagene
[9]:
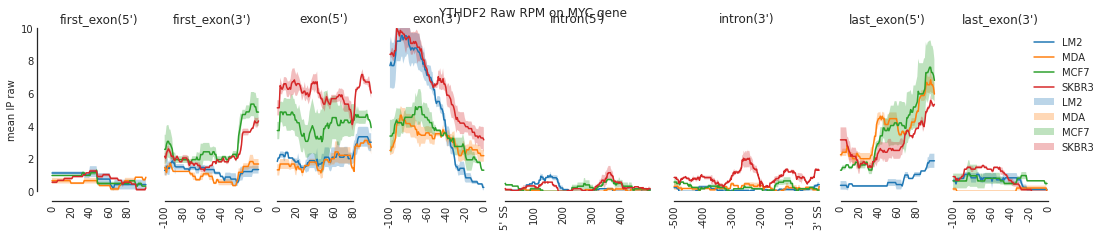
f = plot_mean_density(all_meta_raw, features_to_show = generic_rna, ymax = 10)
plt.suptitle('YTHDF2 Raw RPM on MYC gene')
f=beautify(f)
[ ]: