Using Metadensity with skipper outputs#
Skipper is a new eCLIP pipeline that is faster and more sensitive.
Technically, Metadensity does not care how the bigwig files/bed/bams files are generated. Therefore, whether you process the data with skipper, or ENCODE pipeline, or whatever pipeline you wrote, as long as the file formats (.bigwig, .bam, .bed) are right, Metadensity runs.
This notebook offers a shortcut for users that finds python annoying to copy paste from.
[1]:
# set up files associated with each genome coordinates
import metadensity as md
md.settings.from_config_file('/tscc/nfs/home/hsher/Metadensity/config/hg38-tscc2.ini')
# then import the modules
from metadensity.metadensity import *
from metadensity.plotd import *
import pandas as pd
import matplotlib.pyplot as plt
%matplotlib inline
# I have a precompiles list of ENCODE datas as a csv that loads in this dataloader
plt.style.use('seaborn-white')
please set the right config according to genome coordinate
Using /home/hsher/gencode_coords/GRCh38.p13.genome.fa
Using HG38 by default
Using /tscc/nfs/home/hsher/gencode_coords/GRCh38.p13.genome.fa
Matplotlib created a temporary config/cache directory at /tmp/matplotlib-z0irv727 because the default path (/home/jovyan/.cache/matplotlib) is not a writable directory; it is highly recommended to set the MPLCONFIGDIR environment variable to a writable directory, in particular to speed up the import of Matplotlib and to better support multiprocessing.
Using: /tscc/nfs/home/hsher/gencode_coords/gencode.v33.transcript.gff3
load RBPs into eCLIP object from Skipper outputs#
Notice the bigwig files skipper produce are ‘read truncation counts’, which is different from the coverage track from encode pipeline. Therefore, it is sparser than read coverage. the plot looks more noisy than coverage bigwig tracks. To calculate coverage tracks instead, you can make use of rules in here
[2]:
from pathlib import Path
skipper_output_dir = Path('/tscc/projects/ps-yeolab5/encore/processing/skipper_output/')
[3]:
transcript.fn
[3]:
'/tscc/nfs/home/hsher/gencode_coords/gencode.v33.transcript.gff3'
[4]:
def get_file_path(prefix, skipper_output_dir, ip_suffix = '_IP_',
in_suffix = '_IN_', rep = 1):
return pd.Series({'uid':prefix+f'_rep{rep}',
'RBP': prefix+f'_rep{rep}',
'bam_0': str(skipper_output_dir/'bams'/'dedup'/'genome'/f'{prefix}{ip_suffix}{rep}.genome.Aligned.sort.dedup.bam'),
'bam_control_0':str(skipper_output_dir/'bams'/'dedup'/'genome'/f'{prefix}{in_suffix}{rep}.genome.Aligned.sort.dedup.bam'),
'minus_0': str(skipper_output_dir / 'bigwigs/unscaled' / 'minus' / f'{prefix}{ip_suffix}{rep}.unscaled.minus.bw'),
'minus_control_0': str(skipper_output_dir / 'bigwigs/unscaled' / 'minus' / f'{prefix}{in_suffix}{rep}.unscaled.minus.bw'),
'plus_0': str(skipper_output_dir / 'bigwigs/unscaled' / 'plus' / f'{prefix}{ip_suffix}{rep}.unscaled.plus.bw'),
'plus_control_0': str(skipper_output_dir / 'bigwigs/unscaled' / 'plus' / f'{prefix}{in_suffix}{rep}.unscaled.plus.bw'),
'bed_0': transcript.fn # This is not being used as the later function select transcripts.
#OBSOLETE. newer version will not require this
# function `find_gene_ids_with_reproducible_windows` dictates what genes to be used
}
)
[5]:
# check if files exist.
# if your bigwigs don't exist, need to uncomment the rule all output bigwig/ line
get_file_path('4121', skipper_output_dir).apply(os.path.isfile)
[5]:
uid False
RBP False
bam_0 True
bam_control_0 True
minus_0 True
minus_control_0 True
plus_0 True
plus_control_0 True
bed_0 True
dtype: bool
[6]:
e = eCLIP.from_series(get_file_path('4212', skipper_output_dir))
The reason I did not put enriched windows into bed_* and idr_ is because their format is not really a bed file#
and that will crash Metadensity, as it is using BedTool. alternatively, I would use transcripts= kwargs to specify the transcripts containing reproducible windows
[7]:
from itertools import chain
def find_gene_ids_with_reproducible_windows(prefix, skipper_output_dir):
window_path = skipper_output_dir / 'reproducible_enriched_windows' / f'{prefix}.reproducible_enriched_windows.tsv.gz'
window = pd.read_csv(window_path, sep = '\t')
# find genes with reproducible windows. Some windows contain >1 gene id. `chain` flattens the list of list
# chain https://www.geeksforgeeks.org/python-itertools-chain/
gene_with_windows = list(set(chain(*window['gene_id'].apply(lambda idlist: idlist.split(':')).tolist())))
# skipper is run with gencode v38 which is different from metadensity's version. need some conversion
# remove version
geneid_no_version = [g.split('.')[0] for g in gene_with_windows] # this is list comprehension. lazy and short for loops
# map to Metadensity's version
metadensity_gene_ids = [t.attrs['gene_id'] for t in transcript if t.attrs['gene_id'].split('.')[0] in geneid_no_version]
return metadensity_gene_ids
[8]:
geneids = find_gene_ids_with_reproducible_windows('SNRNP200_HepG2', skipper_output_dir)
[9]:
# here for the set of transcript, we use the IDR peak containing transcript assuming they have good signal
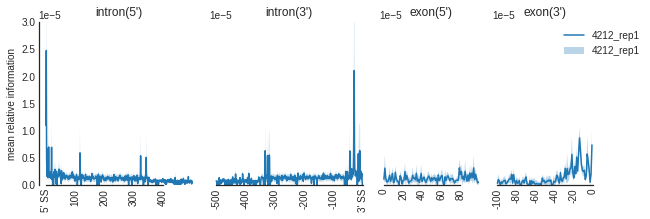
m = Metadensity(e, e.name,background_method = 'relative information', normalize = False, transcript_ids = geneids)
m.get_density_array()
Using: /tscc/nfs/home/hsher/projects/Metadensity/metadensity/data/hg38/gencode
Done building metagene
[10]:
f=plot_mean_density([m],
features_to_show = ['intron', 'exon'], ymax = 0.00003)
f=beautify(f, offset = 0) # sns.despine
f.get_axes()[0].set_ylabel('mean relative information')
[10]:
Text(0, 0.5, 'mean relative information')
[ ]: