Analysis of intronic polyA sites#
This notebook demostrates on how to create cutomized features. The examples is using intronic polyA annotations from the polyADB
[1]:
# set up files associated with each genome coordinates
import metadensity as md
md.settings.from_config_file('/tscc/nfs/home/hsher/Metadensity/config/hg38-tscc2.ini')
# then import the modules
from metadensity.metadensity import *
from metadensity.plotd import *
import pandas as pd
import matplotlib.pyplot as plt
# I have a precompiles list of ENCODE datas as a csv that loads in this dataloader
import sys
sys.path.append('/home/hsher/Metadensity/scripts')
from dataloader import *
%matplotlib inline
Using /home/hsher/gencode_coords/GRCh38.p13.genome.fa
using /home/hsher/gencode_coords/GRCh38.p13.genome.fa
Using: /home/hsher/gencode_coords/gencode.v33.transcript.gff3
[2]:
# use katie's PCF11
katie_data = pd.read_csv('/home/hsher/projects/katie/katie_eclip.csv', index_col = 0)
[3]:
# show a row of the csv file.
master_df.loc[master_df['RBP']=='CPSF6']
[3]:
| uid | RBP | Cell line | bam_0 | bam_1 | bam_control | minus_0 | minus_1 | minus_control | plus_0 | ... | bed_0 | bed_1 | Batch | prefix | bam_control_0 | bam_control_1 | plus_control_0 | plus_control_1 | minus_control_0 | minus_control_1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 28 | 258 | CPSF6 | K562 | /projects/ps-yeolab3/encode/analysis/encode_GR... | /projects/ps-yeolab3/encode/analysis/encode_GR... | /projects/ps-yeolab3/encode/analysis/encode_GR... | /projects/ps-yeolab3/encode/analysis/encode_GR... | /projects/ps-yeolab3/encode/analysis/encode_GR... | /projects/ps-yeolab3/encode/analysis/encode_GR... | /projects/ps-yeolab3/encode/analysis/encode_GR... | ... | /projects/ps-yeolab5/encode/EVN_eCLIP_analysis... | /projects/ps-yeolab5/encode/EVN_eCLIP_analysis... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
1 rows × 23 columns
[4]:
# show a row of the csv file.
katie_data.loc[katie_data['uid']=='PCF11']
[4]:
| bam_0 | bam_control_0 | minus_0 | minus_control_0 | plus_0 | plus_control_0 | bed_0 | bam_1 | bam_control_1 | minus_1 | minus_control_1 | plus_1 | plus_control_1 | bed_1 | uid | RBP | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | /home/wjin/projects/CLIP_seq/Kris_Katie/Data/P... | PCF11 | PCF11 |
[5]:
# load each row the data file into an eCLIP Object
U2AF1 = eCLIP.from_series(encode_data.loc[(encode_data['RBP'] == 'U2AF1')&(encode_data['Cell line'] == 'HepG2')].iloc[0], single_end = False)
U2AF2 = eCLIP.from_series(encode_data.loc[(encode_data['RBP'] == 'U2AF2')&(encode_data['Cell line'] == 'HepG2')].iloc[0], single_end = False)
SF3B4 = eCLIP.from_series(encode_data.loc[(encode_data['RBP'] == 'SF3B4')&(encode_data['Cell line'] == 'HepG2')].iloc[0], single_end = False)
NONO = eCLIP.from_series(encode4_data.loc[(encode4_data['RBP'] == 'NONO')&(encode4_data['Cell line'] == 'HepG2')].iloc[0], single_end = True)
CPSF6 = eCLIP.from_series(encode_data.loc[(encode_data['RBP'] == 'CPSF6')&(encode_data['Cell line'] == 'K562')].iloc[0], single_end = False)
PCF11 = eCLIP.from_series(katie_data.loc[katie_data['uid']=='PCF11'].iloc[0], single_end = True)
all_clips = [U2AF1,U2AF2,SF3B4, NONO,CPSF6, PCF11]
Creating custom features on metagene, using intronic polyA sites as an example#
[45]:
os.path.join(settings.root_dir, settings.polya_fname)
[45]:
'/home/hsher/gencode_coords/polyA.atlas.clusters.2.0.GRCh38.96.bed'
[29]:
# load polyA sites files
# (in order of decreasing priority: TE, terminal exon; EX, exonic; IN, intronic; DS, 1,000 nt downstream of an annotated terminal exon; AE, anti-sense to an exon; AI, anti-sense to an intron; AU, 1,000 nt upstream in anti-sense direction of a transcription start site; IG, intergenic)
polyadf = pd.read_csv(os.path.join(settings.root_dir, settings.polya_fname), sep = '\t', header = None, names = ['chrom', 'start', 'end', 'name', 'average expression', 'strand', 'perc_sample','n_sample', 'avg_tpm', 'polyatype', 'polyasignal'])
# two letter code for the cluster annotation (in order of decreasing priority:
# TE, terminal exon; EX, exonic; IN, intronic; DS, 1,000 nt downstream of an annotated terminal exon; AE, anti-sense to an exon; AI, anti-sense to an intron; AU, 1,000 nt upstream in anti-sense direction of a transcription start site; IG, intergenic)
# the chromosome is listed differently
polyadf['chrom'] = 'chr'+polyadf['chrom'].astype(str)
# filter with TPM at least 0.2
polyadf = polyadf.loc[polyadf['average expression']>0.2]
/home/hsher/miniconda3/envs/metadensity/lib/python3.7/site-packages/IPython/core/interactiveshell.py:3173: DtypeWarning: Columns (0) have mixed types.Specify dtype option on import or set low_memory=False.
interactivity=interactivity, compiler=compiler, result=result)
[16]:
# make type specific annotation
def extract_polyA_signal_coordinates(subset_polyadf):
''' subtracting the coordinate of polyA signal from polya dataframe'''
# extract polya signals
signal_coord = []
for index, row in subset_polyadf.iterrows():
if type(row['polyasignal'])==str:
polyasignals = row['polyasignal'].split(';')
for sig in polyasignals:
motif, rela_pos, obs_pos = sig.split('@')
signal_coord.append([row['chrom'],int(obs_pos),int(obs_pos)+1,
row['name'], row['polyatype'], row['strand']])
# make into bed
polysignal_df = pd.DataFrame(signal_coord,
columns= ['chrom', 'start', 'end', 'name', 'score', 'strand'])
polyasignal_bed = BedTool.from_dataframe(polysignal_df)
return polyasignal_bed
def polyAtype_specific_coords(polyAtype, polyadf = polyadf):
''' create polyA related feature for specific types of polyA
polyAtype can be TE, EX, IN, DS..'''
# filter for specific types
subset_polyadf = polyadf.loc[polyadf['polyatype']==polyAtype]
# create bed of polyA sites
polya_site_bed = BedTool.from_dataframe(subset_polyadf)
polya_signal_bed = extract_polyA_signal_coordinates(subset_polyadf)
return polya_site_bed, polya_signal_bed
[30]:
# extract intronic polyA sites and signal
#information about the poly(A) signal(s) that are present upstream of the poly(A) site,
#including the motif, the location with respect to the cleavage site and the genomic coordinate
polya, polyasignal_bed = polyAtype_specific_coords('IN', polyadf = polyadf)
[31]:
# determine which transcript these are in
header = ['chrom', 'start', 'end', 'name', 'score', 'strand', 'attr']
polya_site_df = polya.intersect(transcript,s = True, wb = True).to_dataframe(header = None)[[0,1,2,3,4,5,19]]
polysignal_df = polyasignal_bed.intersect(transcript, s = True, wb = True).to_dataframe(header = None)[[0,1,2,3,4,5,14]]
polya_site_df.columns = header
polysignal_df.columns = header
/home/hsher/miniconda3/envs/metadensity/lib/python3.7/site-packages/pybedtools-0.8.1-py3.7-linux-x86_64.egg/pybedtools/bedtool.py:3681: UserWarning: Default names for filetype bed are:
['chrom', 'start', 'end', 'name', 'score', 'strand', 'thickStart', 'thickEnd', 'itemRgb', 'blockCount', 'blockSizes', 'blockStarts']
but file has 20 fields; you can supply custom names with the `names` kwarg
"`names` kwarg" % (self.file_type, _names, self.field_count())
/home/hsher/miniconda3/envs/metadensity/lib/python3.7/site-packages/pybedtools-0.8.1-py3.7-linux-x86_64.egg/pybedtools/bedtool.py:3681: UserWarning: Default names for filetype bed are:
['chrom', 'start', 'end', 'name', 'score', 'strand', 'thickStart', 'thickEnd', 'itemRgb', 'blockCount', 'blockSizes', 'blockStarts']
but file has 15 fields; you can supply custom names with the `names` kwarg
"`names` kwarg" % (self.file_type, _names, self.field_count())
[34]:
# extract transcript name
def segment_transcript_name(s):
return s.split(';')[0].split('=')[1]
polysignal_df['transcript_id']= polysignal_df['attr'].map(segment_transcript_name)
polya_site_df['transcript_id']= polya_site_df['attr'].map(segment_transcript_name)
Building Metagene objects containing intronic polyA sites#
Now we know where these sites are, and we know what transcripts they are in. The next step is to add the polyA site features into metagene objects.
[35]:
# build metagene from highly expressed in HepG2 cell lines
from metadensity.pos_enrich import *
high_exp_meta = highly_exp_biogps('HEPG2')
/home/hsher/miniconda3/envs/metadensity/lib/python3.7/site-packages/IPython/core/interactiveshell.py:3364: DtypeWarning: Columns (2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110) have mixed types.Specify dtype option on import or set low_memory=False.
if (await self.run_code(code, result, async_=asy)):
Using: /home/hsher/Metadensity/metadensity/data/hg38/gencode
Done building metagene
[36]:
# check how many of those highly expressed contain intronic polya sites
polya_site_df.loc[polya_site_df['transcript_id'].isin(high_exp_meta.keys())].shape
[36]:
(51, 8)
[37]:
# add feature to those metagene
length = 200
for transcript_id in high_exp_meta.keys():
metagene = high_exp_meta[transcript_id]
# add feature
sub = polya_site_df.loc[polya_site_df['transcript_id']==transcript_id]
for index, row in sub.iterrows():
metagene.create_feature(interval = row['start'], feature_name = 'intron_polyAsite',
length = 200
) # POINT FEATURE
sub = polysignal_df.loc[polysignal_df['transcript_id']==transcript_id]
for index, row in sub.iterrows():
metagene.create_feature(interval = row['start'], feature_name = 'intron_polyAsig',
length = 200)
[38]:
# remove metagene that has no intronic polya
for transcript_id in list(high_exp_meta.keys()).copy():
metagene = high_exp_meta[transcript_id]
if 'intron_polyAsite' not in metagene.featnames or 'intron_polyAsig' not in metagene.featnames:
del high_exp_meta[transcript_id]
[39]:
# we get a total of 147 transcripts with annotation
len(high_exp_meta)
[39]:
34
extract density from eCLIP#
The Metadensity Object takes in eCLIP and the metagene we just built with polyA sites, and fetch density around/within each features
[40]:
def build_idr_metadensity(eCLIP, metagene = high_exp_meta):
''' build metadensity object given eCLIP data and metagene that is build with intronic polyA sites'''
m = Metadensity(eCLIP, eCLIP.name, metagenes = metagene,
background_method = 'relative information', normalize = False)
m.featnames = ['intron_polyAsig','intron_polyAsite'] # to restrict only computing for these 2 features
m.get_density_array()
return m
[41]:
# do it for all eCLIP objects
all_meta = [build_idr_metadensity(e) for e in all_clips]
[62]:
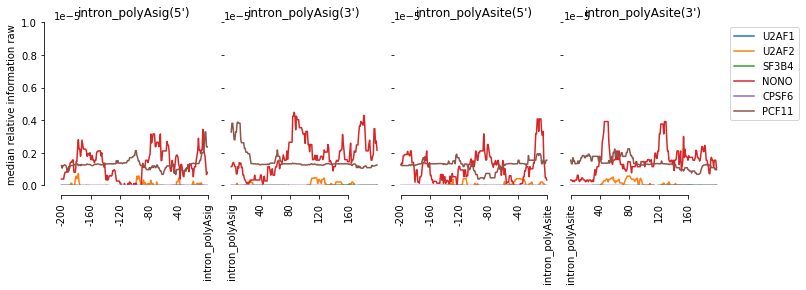
# visualize it
f=plot_mean_density(all_meta,
features_to_show = ['intron_polyAsig','intron_polyAsite'],
ymax = 0.00001, stat = 'median',plot_std = False)
f=beautify(f)
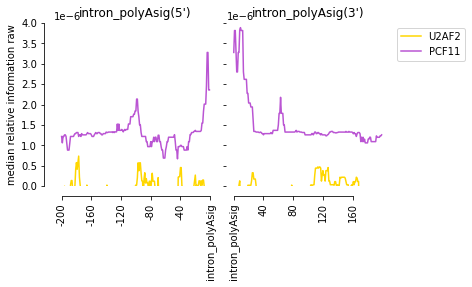
[61]:
f=plot_mean_density([m for m in all_meta if m.name=='PCF11'
or m.name == 'U2AF2'],
features_to_show = ['intron_polyAsig'],
ymax = 0.000004, stat = 'median', plot_std = False,
color_dict = {'PCF11':'mediumorchid', 'U2AF2':'gold'})
f=beautify(f)
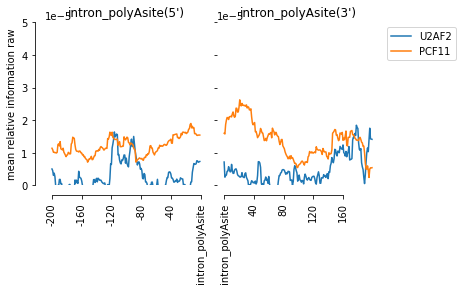
[56]:
f=plot_mean_density([m for m in all_meta if m.name=='PCF11' or m.name == 'U2AF2'],
features_to_show = ['intron_polyAsite'],
ymax = 0.00005, stat = 'mean', plot_std = False)
f=beautify(f)
[19]:
all_meta[0].metagene['ENST00000377577.10'].features['intron_polyAsite']
[19]:
{6647112,
6649008,
6652407,
6653154,
6672178,
6674099,
6691311,
6692044,
6693824,
6700965}
[20]:
settings.point_feature_len
[20]:
50
[21]:
settings.feat_len
[21]:
defaultdict(<function metadensity.config.MetadensityConfig.__init__.<locals>.<lambda>()>,
{'five_prime_UTR': 100,
'three_prime_UTR': 150,
'intron': 500,
'exon': 100,
'first_exon': 100,
'last_exon': 100,
'CDS': 100,
'first_CDS': 100,
'last_CDS': 100,
'3p_duplex': 20,
'5p_duplex': 20,
'mature': 20,
'hairpin': 10,
'pri_mir': 500,
'start_codon': 3,
'stop_codon': 3,
'branchpoint': 50,
'branchpoint_pred': 50,
'full_CDS': 200,
'intron_polyAsite': 200,
'intron_polyAsig': 200})
[30]:
m=all_meta[0].metagene['ENST00000377577.10']
m.features.keys()
[30]:
dict_keys(['exon', 'CDS', 'start_codon', 'stop_codon', 'five_prime_UTR', 'three_prime_UTR', 'intron', 'branchpoint', 'branchpoint_pred', 'last_exon', 'first_exon', 'last_CDS', 'first_CDS', 'intron_polyAsite', 'intron_polyAsig'])
[23]:
len(m.densities['282']['rep1']['intron_polyAsite'])
[23]:
400
[ ]: